
Translate this page into:
Study on mRNA expression of Cajal body – Gemini of coiled body proteins in head and neck squamous cell carcinoma

*Corresponding author: Rooban Thavarajah, Department of Oral and Maxillofacial Pathology, Marundeeswara Oral Pathology Services and Analytics, Chennai - 600 119, Tamil Nadu, India. t.roobanmds@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Thavarajah R, Joseph I, Joshua E, Rao UK, Ranganathan K. Study on mRNA expression of Cajal body – Gemini of coiled body proteins in head and neck squamous cell carcinoma. Int J Mol Immuno Oncol 2020;5(3):108-16.
Abstract
Objectives:
The role of proteins of Cajal bodies (CB) and its identical twin, Gemini of coiled bodies (GEMs) in maintaining genomic integrity and its influence on the initiation, progression, and prognosis of head and neck squamous cell carcinoma (HNSCC) is gaining attention. We attempted to identify the CB and GEM-associated proteins (CB-GEMs) expression in HNSCC patients and study the influence of gender, TP53 mutation, age, and tobacco use on such expression.
Material and Methods:
TP53 mutation, tobacco use, gender, and mRNA levels of CB-GEM proteins of 520 HNSCC cases were collected and subjected to differential expression (DE) analysis. The resultant DE genes were used to create a transcriptional factor gene network using encode chip sequential data. Pathway analysis of the network was performed and presented. P ≤ 0.05 was taken as significant.
Results:
For smoking, the genes GEMIN8, FMR1, TRIM22, and FBL emerged as significantly DE genes. For gender, EAF1, GEMIN8, ZC3H8, TRIM22, FBL, LSG1, ZNF473, GMNC, GEMIN2, ISG20, Opa interacting protein 5, GMNN, and CDK2 were DE gene with statistical significance. For TP53, 15 genes were DE with statistical significance. Transcriptional misregulation in cancer was the frequently affected pathway. The CB-GEM bodies are effective highly conserved, splicesomal organelles that are needed for proper mRNA assembly. Certain mRNA of proteins of the CB-GEM bodies is influenced by TP53 status, gender, and tobacco use.
Conclusion:
The DE of CB-GEM bodies related protein in HNSCC patients are presented. Furthermore, we identified certain critical pathways, where the DE genes of CB-GEM bodies exert critical influence on HNSCC characteristics. This could potentially alter the HNSCC progression, treatment response, and prognosis.
Keywords
Cajal bodies
Gemini of coiled bodies
Spliceosomes
Head and neck squamous cell carcinoma

INTRODUCTION
Eukaryotic cells have evolved with specialized nuclear compartmentalization, called nuclear bodies or subcompartments, to aid cellular functions. They include the nucleoli, Cajal bodies (CBs), Gemini of coiled bodies (GEMs), histone locus bodies, splicing speckles, paraspeckles, as well as nuclear stress bodies.[1-6] CBs, identified by Cajal in 1903, have coilin as their signature protein. They are associated with the processing, assembly, modification and final maturation of spliceosomal small nuclear ribonucleic particles, small nucleolar ribonucleic particles, biogenesis of telomerase, and pre-mRNA assembly. They also contain other type of ribonucleic proteins such as small CB-specific RNPs and small nuclear ribonucleicRNA (snRNA). CB regulates the gene expression through snRNP biogenesis, post-transcriptional modification, snRNA transcription, and assembly. They also play an important role in telomere biology.[3-5]
A nuclear body is similar to CB in morphology, numbers, metabolism, and cell cycle behavior, but devoid of coilin was reported by Liu et al., in 1996. It was much similar and appeared to be twin of CB, so it was named the Gemini of Cajal body (also known as GEM). It is made up of survival motor neuron (SMN) protein complex and other proteins. GEMs are also involved in spliceosomal snRNP biogenesis, transcription, and translation.[1,2,6]
Carcinogenesis is due to orchestrated genomic instability leading to aberrant proliferative signaling, evasive of growth inhibitors, resistant to apoptosis, replicative immortality, neo-angiogenesis, metastasis-modulated inflammation, evasion of immune surveillance, and redirected energy metabolism.[7] Recently, it has been shown that the some CB- associated proteins interact with TP53, protect the genomic integrity, and regulate cell survival. There is a renewed interest in CB due to its role in carcinogenesis and pre- mRNA splicing.[8-11] The association of one such CB protein, WRAP53β, is being increasingly reported in head and neck squamous cell carcinoma (HNSCC), especially with respect to metastasis and treatment outcomes.[12-14] The association of the HNSCC with the CB-GEMS protein(s) alteration remains largely unexplored. These manuscripts aim to identify the CB and GEM-associated proteins (CB-GEMs) expression in HNSCC patients and study the influence of gender, TP53 mutation, age, and tobacco use on such expression.
MATERIAL AND METHODS
The genes associated with CB-GEM bodies were collected from gene ontology site with accession numbers GO:0015030 and GO:0097504, respectively.[15] In addition, the literature scan was performed using Google scholar to identify other associated, reported proteins. The search terms were “CB” “Gemini of CBs,” “GEMs” “Proteins” in published peer- reviewed English literature with no specified time limit. From such sources, the list of proteins associated with the CB-GEM was listed and used for this study.[1-6] Using this list of CB-GB genes, a principal component analysis (PCA) was performed to check the difference in the expression of CB-GB genes in HNSCC and normal tissues using Gene Expression Profiling Interactive Analysis (GEPIA) platform with the Cancer Genome Atlas (TCGA) database.[16]
We collected the existing HNSCC lesional mRNA expression data of CB-GEM-associated proteins from previously published HNSCC studies from Cancer Genome Atlas through the www.cBioportal.com.[17,18] Only patients that had data pertaining to age, gender, tobacco habits, and TP53 mutation were included for the study. Tobacco use was classified as never-user/ever-user, age (≥61; ≤60 years), was downloaded along with TP53 mutation status (present/ absent).
Using the www.NetworkAnalyst.ca (a visual analytics platform for comprehensive gene expression profiling and meta-analysis), the raw data were then analyzed for differential expression (DE) using a statistical, visual, and network-based approach for meta-analysis of expression genetic data (Version-3; last updated July 30, 2019).[19] The single gene expression table mode was used. The variance filter was set maximum permissible limit of 50 (maximum, to remove the data that appear to be less informative or erroneous) and low abundance at 40 (to filter off the data with count lower than a threshold set) was used. Data normalization was done using Log2counts per million. DE analysis was performed with TP53 status, age, gender, and smoking habit as the primary factor with Limma statistics.
For reliable detection of mRNA transcriptional differences and ensure uniform count distribution, continuous mRNA data were normalized using log2counts per million. We approached with TP53/age/gender/smoking status as a primary factor alone. P ≤ 0.05 and data presented along with Log Fold Change (LFC) values. The differentially expressed genes (DEGs) for TP53 status, age, gender, and smoking status are presented. LFC threshold was preserved at 0, and actual obtained values quoted along with P-value, as there are issues with regard to LFC. LFC is one of the measures of change in the expression level of genes. It inherently carries a disadvantage of being biased and misclassified. It would miss the DE entities with large differences but smaller ratios, leading to inaccurate identification of changes, especially high expression levels.[20]
An attempt was made to identify the transcription factor (TF) network of association of DEG that is presented. Encode chip sequential data were used to plot the TF network.[21] Each of the resultant networks had a node (visual representation of DE gene/protein), edge (visual representation of a relation, line that connect two nodes), and a seed (DE gene/protein). Besides the genes, the default settings of degree (number of connections that a node has with other nodes) and the betweenness (number of shortest path, through a particular node) were used.[19] If the network was large, minimum network option was used. Kyoto Encyclopedia of Genes and Genomes (KEGG)[22] pathway enrichment analysis was performed to identify significant pathways along with the total number of genes in pathway, number of genes in the depicted pathway, P-value and false detection rate are presented.
RESULTS
We identified 60 genes for CB and 11 genes for GEM from the gene ontology website. The literature search yielded ten more genes. There were 81 genes associated with the CB-GEM that regulates or is associated with them. We subjected these gene list, in official gene nomenclature with GEPIA 2 data interface to analyze the DE of these genes, between normal (n = 44) and HNSCC lesional tissues (n = 519), using PCA and employing the TCGA-HNSCC database. We identified that there was clustering of the genes in a remarkable fashion, confirming the difference in expression of select genes [Figure 1]. From the www.cbioportal.org, web portal, the details of mRNA expression, demographic characters were obtained. Furthermore, the same portal was used to identify any genetic alterations (copy number alteration/ mutation/altered mRNA expression (±2 standard deviation), which was collated for 488 patients. The distribution across demographics is shown in Figure 2. Of the 488 patients, 324 (62.3%) had at least one genetic alteration. The study population was also classified as with and without alterations.
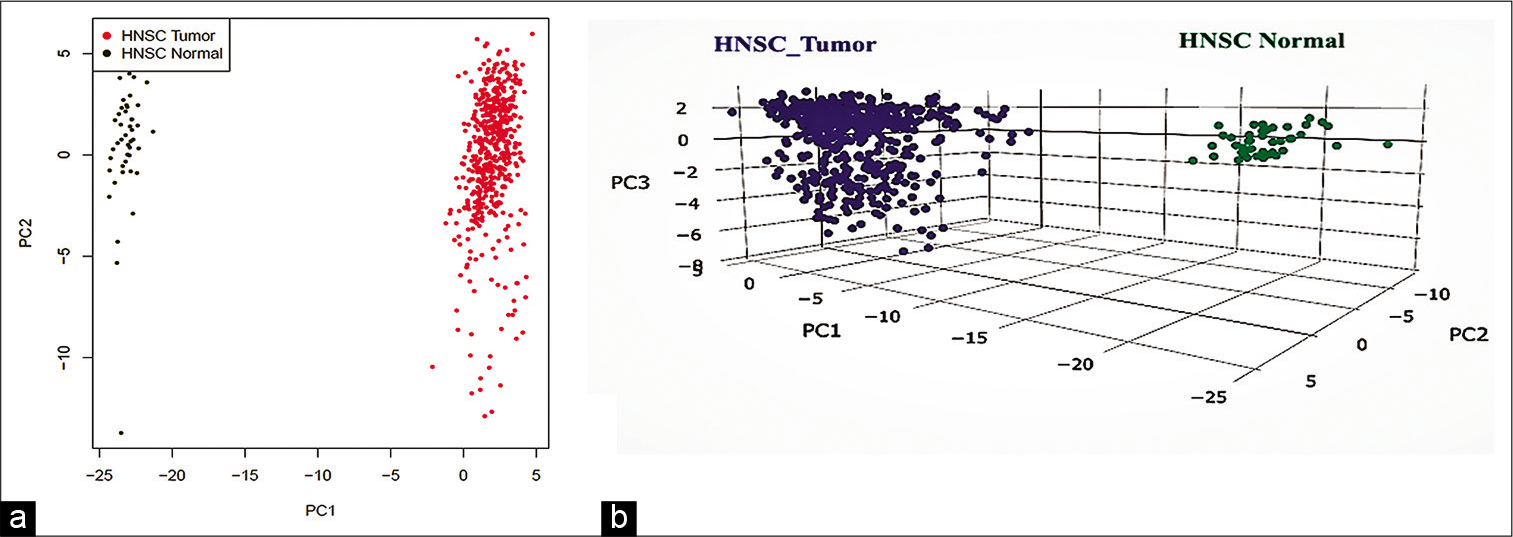
- The principal component analysis of mRNA of the Cajal body – Gemini of coiled bodies proteins in the Cancer Genome Atlas – head and neck squamous cell carcinoma lesional and normal tissue. (a) Two-dimensional plot of principal component analysis (PCA) plot, (b) three-dimensional plot of PCA, blue tumor, green normal.
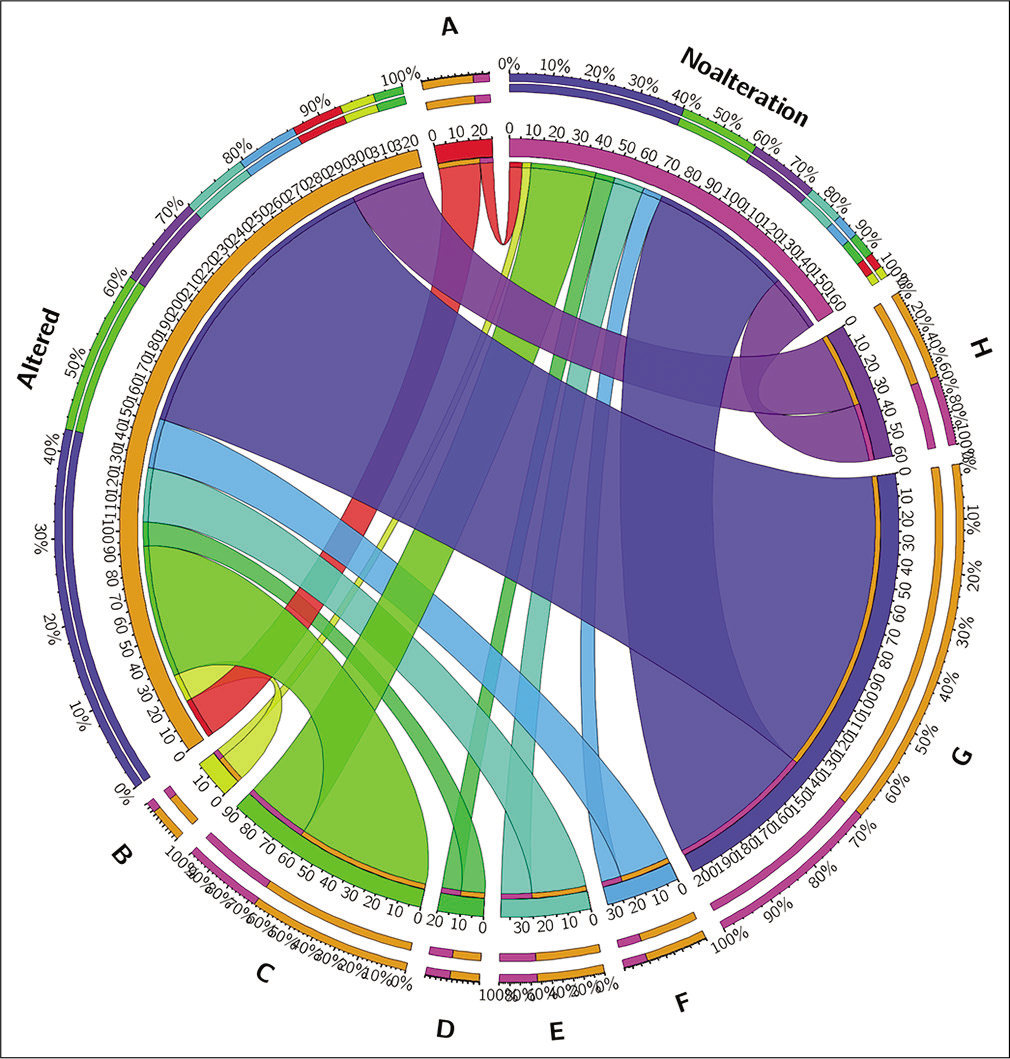
- Alteration – Genetic/copy number alterations/altered mRNA expression (±2 standard deviation); no alterations – No genetic/copy number alterations/altered mRNA expression (±2 standard deviation). (a) – Wild TP53, non-smoker, male; (b) – wild TP53, non-smoker, female; (c) – wild TP53, ever-smoker, male; (d) – wild TP53, ever-smoker, female; (e) – mutated TP53, non- smoker, male; (f) – mutated TP53, non-smoker, female; (g) – mutated TP53, ever-smoker, male; (h) – mutated TP53, ever-smoker, female.
Single table gene DE was carried out using the www.Networkanalyst.ca. During the initial process, the CCT2, DDX42, and EFTUD2 were removed and 78 genes processed. During the normalization process, no gene was removed on account of low count, while 40 genes removed for low abundance.
When, TP53 was used as a primary factor, 18 different genes emerged as DE with significance [Table 1]. The subsequent TF-gene interaction network had 252 nodes, 562 edges, and 18 seeds, which was minimized to 67 nodes, 210 edges from 18 seeds [Figure 3]. The KEGG pathway enrichment analysis showed that several pathways were affected [Table 2]. When smoking was employed as primary factor, only four mRNAs of genes of RuvB such as AAA ATPase 1 (RUVBL1) (LFC = 0.17668; P = 0.016), GEMIN8 (LFC = 0.20484;P = 0.023), TRIM22 (LFC = −0.47611;P = 0.025), and FBL (LFC = 0.21427;P = 0.028) emerged significant. The network analysis showed that 74 nodes and 74 edges were formed with these as seeds [Figure 3]. The role of GABPA, MLLT1, and KLF9 was identified. The KEGG pathway analysis revealed that transcriptional misregulation in cancer, cell cycle, and transforming growth factor (TGF)-beta signaling pathway was notably involved as compared with other pathways [Table 2]. When gender was used the main factor, there were 14 genes differentially expressed. The TF-gene network revealed a 195 node, 345 edge network from 12 seed one, which when minimized, formed a network made up of 44 node, 100 edge from 12 seeds. The role of CDK2 was also identified [Figure 3]. The KEGG pathway analysis revealed few cancer-related pathways involvement [Table 2]. In the age group, there were no genes that had significant DE.
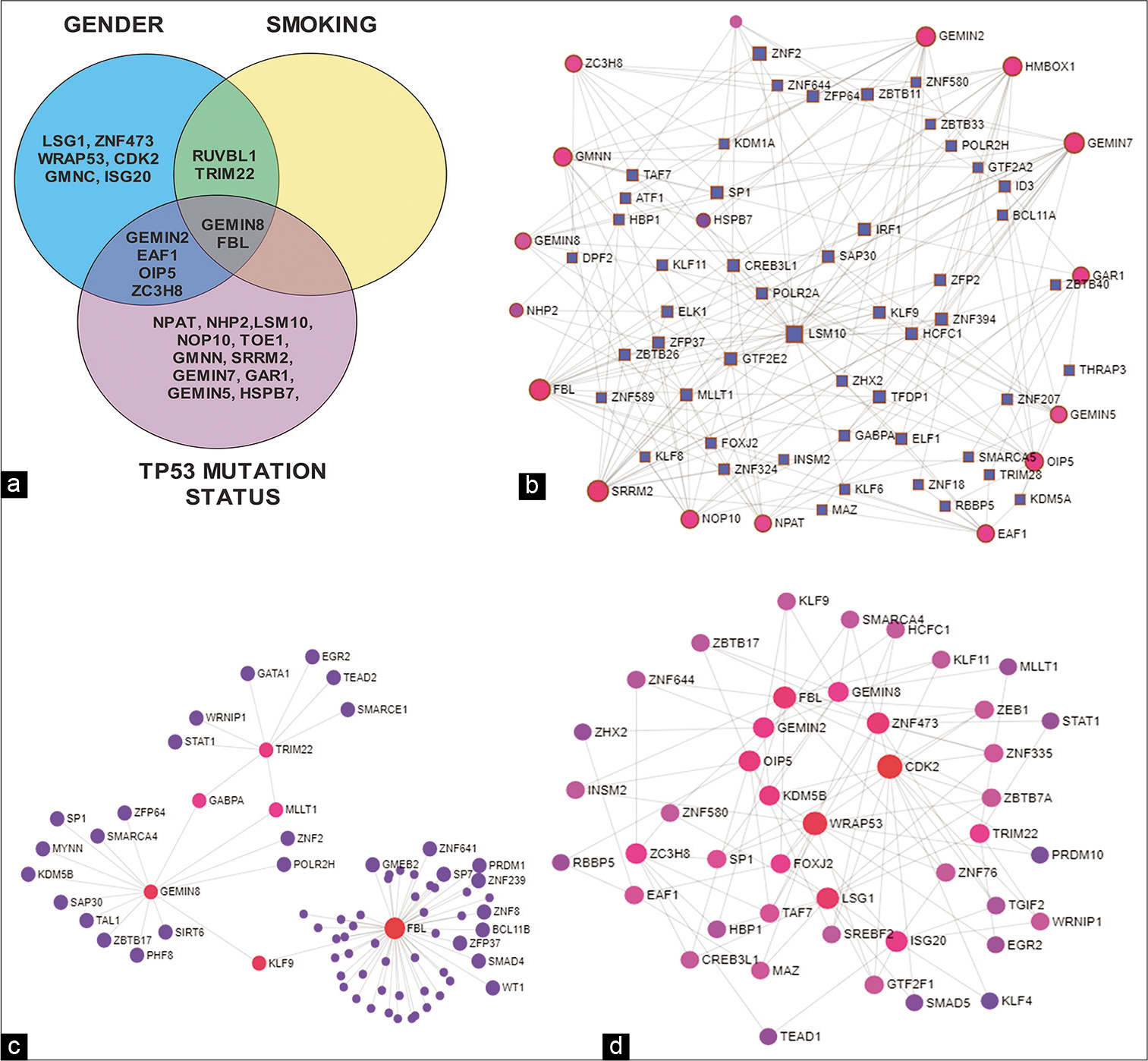
- (a) – Venn diagram showing the differential expression (DE) genes of Cajal body – Gemini of coiled bodies (CB-GEM) proteins between TP53 mutation, gender, and smoking status; (b) – network diagram of transcription factor – Gene interaction of DE genes in CB-GEM in TP53 mutation status; (c) – network diagram of transcription factor – gene interaction of DE genes in CB-GEM in Smoking; (d) – network diagram of transcription factor – gene interaction of DE genes in CB-GEM on gender.
| TP53 status | mRNA | Mean±SD | Mean±SD | Differential expression | ||
|---|---|---|---|---|---|---|
| Symbol | Wild TP53 | Mutant TP53 | logFC | P-value | Adjusted P-value | |
| GAR1 | 567.57±232.39 | 424.81±178.26 | −0.34132 | 1.01E-12 | 3.85E-11 | |
| HMBOX1 | 78.98±54.35 | 99.9±54.48 | 0.47493 | 2.22E-09 | 4.22E-08 | |
| GEMIN5 | 572.32±214.94 | 654.8±238.05 | 0.28208 | 9.24E-09 | 1.17E-07 | |
| GMNN | 647.07±352.13 | 497.01±288.95 | −0.30821 | 1.95E-07 | 1.85E-06 | |
| EAF1 | 706.03±327.17 | 820.63±342.85 | 0.32647 | 4.67E-07 | 3.13E-06 | |
| NPAT | 365.07±182.31 | 428.49±176.61 | 0.33757 | 4.94E-07 | 3.13E-06 | |
| GEMIN7 | 413.42±197.8 | 334.25±200.83 | −0.23758 | 6.82E-06 | 3.70E-05 | |
| NHP2 | 1639.64±1109.06 | 1248.02±597.81 | −0.22257 | 7.95E-05 | 0.000378 | |
| OIP5 | 177.74±97.47 | 145.05±106.68 | −0.27156 | 0.000236 | 0.000997 | |
| TOE1 | 275.54±106.96 | 231.33±86.86 | −0.16783 | 0.000289 | 0.001099 | |
| SRRM2 | 8735.89±3041.7 | 9493.13±3157.75 | 0.20128 | 0.000423 | 0.00146 | |
| ZC3H8 | 213.29±95.04 | 180.75±83.41 | −0.1722 | 0.001191 | 0.00377 | |
| NOP10 | 1888.57±1084.97 | 1549.29±779.69 | −0.18102 | 0.001788 | 0.005226 | |
| FBL | 4577.02±3304.99 | 3659.35±2431.98 | −0.17835 | 0.006186 | 0.01679 | |
| HSPB7 | 554.76±1935.33 | 710.29±2496.6 | 0.81149 | 0.010378 | 0.026291 | |
| GEMIN2 | 214.99±94.43 | 190.99±116.08 | −0.12867 | 0.012705 | 0.029115 | |
| LSM10 | 600.2±242.92 | 521.13±225.85 | −0.11474 | 0.013025 | 0.029115 | |
| GEMIN8 | 305±129.84 | 261.96±112.63 | −0.14029 | 0.014457 | 0.03052 | |
| Sex | Female | Male | ||||
| EAF1 | 892.17±407 | 747.25±307.41 | 0.30377 | 8.78E-06 | 0.00022 | |
| GEMIN8 | 305.08±135.54 | 264.73±111.96 | 0.26296 | 1.16E-05 | 0.00022 | |
| WRAP53 | 302.15±129.26 | 373.74±205.57 | −0.21695 | 7.82E-05 | 0.000991 | |
| RUVBL1 | 1661.37±715.33 | 1968.55±807.71 | −0.17519 | 0.000235 | 0.00193 | |
| ZC3H8 | 168.04±83.4 | 198.89±88.78 | −0.20183 | 0.0003 | 0.00193 | |
| TRIM22 | 2514.9±2050.38 | 1886.29±1786.01 | 0.52166 | 0.000347 | 0.00193 | |
| FBL | 3217.22±1704.06 | 4200.69±3009.79 | −0.24413 | 0.000355 | 0.00193 | |
| LSG1 | 1341.76±545.44 | 1617.76±865.77 | −0.16957 | 0.002122 | 0.01008 | |
| ZNF473 | 239.52±96.58 | 284.42±123.89 | −0.1624 | 0.002614 | 0.011038 | |
| GMNC | 0.28±0.66 | 1.72±9.56 | −0.33368 | 0.004628 | 0.017585 | |
| OIP5 | 130.28±76.15 | 163.99±112.14 | −0.21167 | 0.006604 | 0.022813 | |
| ISG20 | 947.28±971.9 | 806.13±905.48 | 0.32511 | 0.007455 | 0.023607 | |
| GEMIN2 | 174.27±70.89 | 206.97±120.15 | −0.14276 | 0.008566 | 0.025038 | |
| CDK2 | 875.88±328.63 | 1032.24±423.51 | −0.14276 | 0.009957 | 0.027025 | |
| Smoking status | Non-smoker | Ever-smoker | ||||
| RUVBL1 | 1716.5±838.94 | 1938.06±776.41 | 0.17668 | 0.000427 | 0.016217 | |
| GEMIN8 | 302.48±122.05 | 267.39±118.08 | −0.20484 | 0.001229 | 0.023353 | |
| TRIM22 | 2499.47±2154.44 | 1920.41±1770.48 | −0.47611 | 0.001953 | 0.024734 | |
| FBL | 3602.37±3086.71 | 4042.5±2656.02 | 0.21427 | 0.002953 | 0.028052 |
| Pathway | Total | Network – TP53 mutation | Network – smoking | Gender | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Expected | Hits | P-value | FDR | Expected | Hits | P-value | FDR | Expected | Hits | P-value | FDR | ||
| Transcriptional misregulation in cancer | 186 | 2.6 | 24 | 3.53E-17 | 1.12E-14 | 0.721 | 6 | 6.54E-05 | 0.0208 | 1.92 | 11 | 2.85E-06 | 0.000453 |
| Basal transcription factors | 45 | 0.628 | 6 | 3.37E-05 | 0.00178 | 0.465 | 4 | 0.00114 | 0.0404 | ||||
| TGF-beta signaling pathway | 92 | 1.28 | 8 | 3.91E-05 | 0.00178 | 0.357 | 3 | 0.00524 | 0.333 | 0.951 | 7 | 4.19E-05 | 0.00266 |
| Cell cycle | 124 | 1.73 | 8 | 0.00032 | 0.00926 | 0.481 | 4 | 0.00125 | 0.198 | 1.28 | 7 | 0.000276 | 0.0125 |
| Pathways in cancer | 530 | 7.4 | 18 | 0.00035 | 0.00928 | 2.06 | 6 | 0.0145 | 0.77 | 5.48 | 13 | 0.00284 | 0.0759 |
| Viral carcinogenesis | 201 | 2.81 | 10 | 0.000473 | 0.0108 | 2.08 | 10 | 3.78E-05 | 0.00266 | ||||
| Longevity regulating pathway | 89 | 1.24 | 6 | 0.00146 | 0.0291 | ||||||||
| Cellular senescence | 160 | 2.23 | 8 | 0.00172 | 0.0303 | 1.65 | 5 | 0.0246 | 0.27 | ||||
| TNF signaling pathway | 110 | 1.54 | 6 | 0.00426 | 0.0565 | 1.14 | 5 | 0.00547 | 0.0916 | ||||
| Signaling pathways regulating pluripotency of stem cells | 139 | 1.94 | 6 | 0.0129 | 0.136 | 1.44 | 5 | 0.0143 | 0.197 |
When genetic alterations were used as a primary factor, only ZC3H8 emerged as a DE gene. In no genetic alteration subgroup, it was expressed 171.6 ± 80.99, while in those with alteration subgroup, it was 200.83 ± 91.44. The LFC was 0.2 with adjusted P = 0.00741. No network could not be built with 1 seed. We attempted to repeat the process using genetic alteration as a secondary blocking factor. For smoking, the genes GEMIN8, FMR1, TRIM22, and FBL emerged as significantly DE genes. For gender, EAF1, GEMIN8, ZC3H8, TRIM22, FBL, LSG1, ZNF473, GMNC, GEMIN2, ISG20, Opa interacting protein 5 (OIP5), GMNN, and CDK2 were DE gene with statistical significance. For TP53, the genes GAR1, HMBOX1, GEMIN5, GMNN, NPAT, EAF1, GEMIN7, GEMIN6, NHP2, SRRM2, TOE1, FRG1, OIP5, NOP10, ZC3H8, LSM10, GEMIN8, GEMIN2, FBL, HSPB7, HINFP, and LSG1 were DE with statistical significance. For all these genes, P ≤ 0.05 and LFC ≤ 1.
DISCUSSION
Genetic instability and alteration are the driving force of carcinogenesis and mRNA splicing and assembly play a crucial role. Most of these events happen in CB- GEM and these bodies could potentially play a key role in carcinogenesis. There are recent reports of CB involvement in the genomic stability and TP53 mutation. There are very few reports of WRAP53β influencing the HNSCC course and outcome.[9-12] To the best of our knowledge, there are no detailed reports of CB-GEM proteins influence on HNSCC. In this manuscript, we attempt to screen for CB-GEM protein mRNA expression in HNSCC and see the influence of TP53 mutation, tobacco exposure, gender, and age.
There was no significant DE of CB-GEM gene mRNAs between the age groups. With regard to the genetic alterations, six in ten HNSCC patients had at least one CB-GEM anomaly. ZC3H8 was the only gene to be DE between these two groups with P = 0.00741. It has been reported that tumor cells with the lower ZC3H8 expression exhibited decreased proliferation rates, slower migration, reduced ability to invade through a basement membrane, and decreased anchorage independent growth in vitro. In the present study, with no genetic alteration in CB-GEM subgroup, ZC3H8 was lower than those with at least one altered CB-GEM mRNAs and those with wild TP53 had higher ZC3H8 concurring with the previous studies on oncogenic phenotypes.[23]
GEMIN8 and FBL were DE among gender, smoking, and TP53 status. GEMIN8 is required for spliceosomal snRNP assembly in the cytoplasm and pre-mRNA splicing in the nucleus. This gene upholds the structural organization of the SMN complex and mediates with other GEMIN proteins for the snRNP assembly. The DE of GEMIN8 mRNA levels is increased in females, non-smokers and those with wild TP53. The higher DE in wild TP53 is in reflection of the role played by GEMIN8 in maintain the SMN complex and thereby the CB-GEM.[24] Till date, the role of GEMIN8 in HNSCC is not explored.
FBL gene is a part of snRNP that mediates the first step in processing preribosomal RNA. It is associated with the U3, U8, and U13 small nuclear RNAs and is located in the dense fibrillar component of the nucleolus and also rarely in other cell organelles. FBL mediates rDNA chromatin, during progression through mitosis. FBL also participates indirectly in normal chromatin dynamics.[25] In our study, FBL levels were higher in wild TP53, where as in breast cancer, FBL mRNA levels were significantly higher in mutant p53 tumors compared to wild-type p53 tumors.[26] FBL is upregulated in males and ever smokers, reflecting their higher risk, and poor prognosis.[27] This difference in expression in HNSCC warrants further studies.
The mRNA of TRIM22 was increased in females and never smokers, as reported previously.[27] TRIM22 activates the non-canonical but not the canonical NF-κB pathway.[28] However, in lung cancer, its overexpression has been associated with advanced tumor, node, metastasis stage, positive nodal metastasis, and poor prognosis. Increased TRIM22 is reported to promote proliferation, colony formation, and invasion in cell lines and depletion reduces the same. TRIM22 overexpression promotes cell cycle progression through regulation of cyclin D1, cyclin E, and p27. TRIM22 also is known to alter E-cadherin N-cadherin, Vimentin, and Snail. Furthermore, TRIM22 activated PI3K/AKT/GSK3β/β-catenin oncogenic signaling pathways.[29] This difference, in terms of HNSCC, warrants further exploration.
RUVBL1 was increased in ever-smokers and males. The protein RUVBL1 relates to the Wnt/Hedgehog/Notch and C-MYC pathway. It mediates the acetylation of nucleosomal histones H4 and H2A. This alters the nucleosome – DNA interactions, particularly the ones that positively regulate transcription. Such transcriptional programs are often associated with oncogene and proto-oncogene-mediated growth induction, tumor suppressor-mediated growth arrest and replicative senescence, apoptosis, and DNA repair.[30,31]
The role of RUVBL1 in HNSCC needs further exploration.
GEMIN2 and OIP5 are highly expressed in males (P = 0.03 and 0.02, respectively) and wild TP53 (P = 0.03 and 0.001, respectively) group with statistical significance. The role of GEMIN2 in carcinogenesis is unknown but in conjunction, works with other GEMIN proteins.[2] The OIP5 is a known marker in carcinogenesis with poor outcomes.[32,33] The reason for this difference remains to be elucidated.
Among genes DE among TP53 subgroup, increase in mRNA levels was noted GAR1, GMNN, GEMIN7, NHP2, TOE1, NOP10, and LSM10 in wild TP53 group. On the contrary, mutant TP53 subgroup exhibited an increase in HMBOX1, GEMIN5, NPAT, SRRM2, and HSPB7. GAR1 is known to downregulate p53 and thereby influences the telomere biology, a critical mortality defining entity.[34] TP53 not only regulates cell cycle progression but also functions through geminin/GMNN to prevent certain gene types amplification and protect genomic integrity.[35] GEMIN7 is known to interfere with the mRNA splicing machinery resulting in activation of the tumor suppressor p53.[36] Hence, in wild TP53, the levels remain high, as identified in the present study. TOE1 participates in cell growth regulation through the upregulation of p21 and a cell cycle delay at the G2/M phase. It also binds to the TP53 modulating its transactivation potential. In our present study, TOE1 levels were higher in wild TP53 group, reflecting this status.[37] The role of these proteins/genes in HNSCC is yet to be studied.
The HMBOX1 is known to downregulate the expression of anti-apoptotic proteins (Bcl-2 and Bcl-xL), raised the expression of pro-apoptotic-regulated proteins (Bad and Bax), apoptotic executioner (Caspase3), and TP53. In the present study, the HMBOX1 levels are increased, as reported earlier.[38] GEMIN5 levels were increased with mutant TP53 and consistent with the previous zebrafish studies indicate that GEMIN5 is associated with TP53 regulation/ activation.[39] Studies have shown that normal TP53 decreases the expression of the NPAT through a G1 cell-cycle arrest. In the present study, in the mutant TP53 group, NPAT levels are increased, that is, consistent with the previous report.[40] Resultant decreased expression of NPAT following p53 activation down decreases CDK9 recruitment. It is also shown that induction of a G1 cell-cycle arrest alters histone mRNA 3’ end processing, which may be altered during tumorigenesis.[40] It has been reported that in kidney HSPB7 expression is regulated by wild-type p53 and as reported, mutation leads to increased HSPB7 expression.[41] Most of the gene’s influence has not been explored in HNSCC and warrants further studies.
The role of WRAP53 in HNSCC progression as well as its close association with TP53 has been extensively studied.[9-12] Increased expression is associated with better survival. In this study, it is increased among males and this needs further investigation. The role of gender in EAF1 expression also needs to be investigated. Zebrafish studies suggest that eaf1/2, as transcription repressors, might act to suppress transactivation of their binding partners, but also directly activate the expression of their own downstream genes as TFs. Furthermore, eaf1 and eaf2 antagonize Wnt/β-catenin signaling pathway.[42] The role of other genes in cancer and HNSCC has to be explored.
The role of other TFs and intermediaries, particularly the TP53 DE gene network analysis, is shown in Figure 3 and Table 2. The KEGG pathway enrichment analysis shows that most of the genes/network nodes and edges have prominent pathways associated with cancer with sufficient statistical significance. Most of these are the zinc finger moiety. In gender-related DE network analysis, the CDK2 emerged as an important factor. It is a well-known HNSCC prognostic marker and has been widely studied.[43] ETS family TF GABPA is suggested as an oncogenic element in several cancers. It is likely involved in activation of cytochrome oxidase expression and nuclear control of mitochondrial function.[44] Another intermediary, MLLT1 is a component of the super elongation complex (SEC), a complex required to increase the catalytic rate of RNA polymerase II transcription by suppressing transient pausing by the polymerase at multiple sites along the DNA.[45] Kruppel-like factor 9 (KLF9), a TF, is critical for the inhibition of growth and development of tumors. KLF9 role is notable in lung and pancreas tumors.[46] The role of these genes in HNSCC remains to be explored.
In the KEGG pathway enrichment analysis, it appears that DE genes of CB-GEMS genes play a pivotal role in influencing transcriptional misregulation in cancer pathway and TGF-beta signaling pathway [Table 2]. Apart from this cellular senescence, TNF signaling pathway and signaling pathways regulating pluripotency of stem cells were correlated with gender and TP53 mutation. This underlines the fact that certain crucial pathways of cancers are influenced by the CB-GEMS, and the TP53 mutation status possibly alters such influences. It is also an interesting observation, within the confines of this data and study, that the DE of genes in association with smoking is shared with TP53 and gender. This observation needs further in-depth study.
The overall results have to be interpreted keeping in mind the fact that the mRNA protein level correlation is often linear but at instances need not necessarily correlate. This could stem from the fact that mRNA translation can be upregulated by RNA binding proteins, translation upregulation by downregulation of miRNA targeting that particular mRNA, the resultant product protein stability may be altered due to post-translational modifications such as phosphorylation, acetylatylation, and glycosylation that is not under the purview of the mRNA and the possibly may be related to the inherent lifetime of the protein, which may accumulate, while the mRNA is normally quick.[47] In addition, the role of site, HPV infection, stage and grade of the HNSCC, aneuploidy status, comutants, effect of other gene mutations, inflammatory mediators, and other potentially confounding factors could influence the outcomes. Future studies need to factor in such details, when studying CB-GEMs in HNSCCs.
CONCLUSION
This study is probably the first to identify the association of CB-GEM proteins with HNSCC and influence of TP53 mutation on the association. The CB-GEM bodies are crucial for vital cellular process including pre-mRNA splicing and vital for proper functioning of spliceosomal apparatus. These structures are one of the most conserved structures. Cancer associated genes and proteins also have to pass through these nuclear subcompartments to ensure their survival and multiplication process. Alteration of CB-GEM bodies- associated proteins could alter the course of the oncologic process, by interfering with proliferation signals, growth, apoptosis, replication, immortality, angiogenesis, metastasis, cancer-associated inflammation, immune surveillance, and tumor associated energy metabolism. Hence, the knowledge of the CB-GEM bodies in HNSCC, when deciphered fully, could help us to draw optimum treatment protocols.
Declaration of patient consent
Patient’s consent not required as patients identity is not disclosed or compromised. All data obtained from open access sources that have been duly cited.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Nuclear bodies: News insights into structure and function. Curr Opin Cell Biol. 2017;46:94-101.
- [CrossRef] [PubMed] [Google Scholar]
- The role of nuclear bodies in gene expression and disease. Biology (Basel). 2013;2:976-1033.
- [CrossRef] [PubMed] [Google Scholar]
- Towards an understanding of regulating Cajal body activity by protein modification. RNA Biol. 2017;14:761-78.
- [CrossRef] [PubMed] [Google Scholar]
- Cajal bodies are linked to genome conformation. Nat Commun. 2016;7:10966.
- [CrossRef] [PubMed] [Google Scholar]
- Signals controlling Cajal body assembly and function. Int J Biochem Cell Biol. 2013;45:1314-7.
- [CrossRef] [PubMed] [Google Scholar]
- Nuclear subcompartments: An overview. Cell Biol Int. 2017;41:2-7.
- [CrossRef] [PubMed] [Google Scholar]
- Hallmarks of cancer: The next generation. Cell. 2011;144:646-74.
- [CrossRef] [PubMed] [Google Scholar]
- Nuclear inclusion bodies of mutant and wild-type p53 in cancer: A hallmark of p53 inactivation and proteostasis remodelling by p53 aggregation. J Pathol. 2017;242:24-38.
- [CrossRef] [PubMed] [Google Scholar]
- On the road with WRAP53ß: Guardian of Cajal bodies and genome integrity. Front Genet. 2015;6:91.
- [CrossRef] [PubMed] [Google Scholar]
- The Cajal body protein WRAP53ß prepares the scene for repair of DNA double-strand breaks by regulating local ubiquitination. Front Mol Biosci. 2019;6:51.
- [CrossRef] [PubMed] [Google Scholar]
- WRAP53 is essential for Cajal body formation and for targeting the survival of motor neuron complex to Cajal bodies. PLoS Biol. 2010;8:e1000521.
- [CrossRef] [PubMed] [Google Scholar]
- Ultrastructural immunolocalization of telomerase and hyaluronate in migrating keratinocytes in a case of oro-pharyngeal squamous cancer. Pathol Res Pract. 2019;215:215-21.
- [CrossRef] [PubMed] [Google Scholar]
- Nuclear expression of WRAP53ß is associated with a positive response to radiotherapy and improved overall survival in patients with head and neck squamous cell carcinoma. Oral Oncol. 2015;51:24-30.
- [CrossRef] [PubMed] [Google Scholar]
- TCAB1: A potential target for diagnosis and therapy of head and neck carcinomas. Mol Cancer. 2014;13:180.
- [CrossRef] [PubMed] [Google Scholar]
- The gene ontology resource: 20 years and still going strong. Nucleic Acids Res. 2019;47:D330-8.
- [CrossRef] [PubMed] [Google Scholar]
- GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019;47:W556-60.
- [CrossRef] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576-82.
- [CrossRef] [PubMed] [Google Scholar]
- The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401-4.
- [CrossRef] [PubMed] [Google Scholar]
- NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019;47:W234-41.
- [CrossRef] [PubMed] [Google Scholar]
- A variable fold change threshold determines significance for expression microarrays. FASEB J. 2003;17:321-3.
- [CrossRef] [PubMed] [Google Scholar]
- Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat Protoc. 2013;8:2502-15.
- [CrossRef] [PubMed] [Google Scholar]
- KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2011;40:D109-14.
- [CrossRef] [PubMed] [Google Scholar]
- Regulation of the oncogenic phenotype by the nuclear body protein ZC3H8. BMC Cancer. 2018;18:759.
- [CrossRef] [PubMed] [Google Scholar]
- Gemin8 is required for the architecture and function of the survival motor neuron complex. J Biol Chem. 2006;281:37009-16.
- [CrossRef] [Google Scholar]
- Stop-and-go: Dynamics of nucleolar transcription during the cell cycle. Epigenet Insights. 2019;12:2516865719849090.
- [CrossRef] [PubMed] [Google Scholar]
- p53 acts as a safeguard of translational control by regulating fibrillarin and rRNA methylation in cancer. Cancer Cell. 2013;24:318-30.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical update on head and neck cancer: Molecular biology and ongoing challenges. Cell Death Dis. 2019;10:540.
- [CrossRef] [PubMed] [Google Scholar]
- TRIM22 can activate the noncanonical NF-κB pathway by affecting IKKα. J Recept Signal Transduct Res. 2015;35:289-94.
- [CrossRef] [PubMed] [Google Scholar]
- TRIM22 confers poor prognosis and promotes epithelial-mesenchymal transition through regulation of AKT/GSK3ß/ß-catenin signaling in non-small cell lung cancer. Oncotarget. 2017;8:62069-80.
- [CrossRef] [PubMed] [Google Scholar]
- Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Mol Cell Biol. 2004;24:1884-96.
- [CrossRef] [PubMed] [Google Scholar]
- The role of pontin and reptin in cellular physiology and cancer etiology. Front Mol Biosci. 2017;4:58.
- [CrossRef] [PubMed] [Google Scholar]
- Long noncoding RNA OIP5-AS1 promotes the progression of oral squamous cell carcinoma via regulating miR-338-3p/NRP1 axis. Biomed Pharmacother. 2019;118:109259.
- [CrossRef] [PubMed] [Google Scholar]
- LncRNA OIP5-AS1 is overexpressed in undifferentiated oral tumors and integrated analysis identifies as a downstream effector of stemness-associated transcription factors. Sci Rep. 2018;8:7018.
- [CrossRef] [PubMed] [Google Scholar]
- p53 downregulates the fanconi anaemia DNA repair pathway. Nat Commun. 2016;7:11091.
- [CrossRef] [PubMed] [Google Scholar]
- Geminin functions downstream of p53 in K-ras-induced gene amplification of dihydrofolate reductase. Cancer Res. 2012;72:6153-62.
- [CrossRef] [PubMed] [Google Scholar]
- p53 is activated in response to disruption of the pre-mRNA splicing machinery. Oncogene. 2013;32:1-14.
- [CrossRef] [PubMed] [Google Scholar]
- TOE1 interacts with p53 to modulate its transactivation potential. FEBS Lett. 2009;583:2165-70.
- [CrossRef] [PubMed] [Google Scholar]
- Low expression level of HMBOX1 in high-grade serous ovarian cancer accelerates cell proliferation by inhibiting cell apoptosis. Biochem Biophys Res Commun. 2018;501:380-6.
- [CrossRef] [PubMed] [Google Scholar]
- A subset of SMN complex members have a specific role in tissue regeneration via ERBB pathway-mediated proliferation. NPJ Regen Med. 2020;5:6.
- [CrossRef] [PubMed] [Google Scholar]
- Induced G1 cell-cycle arrest controls replication-dependent histone mRNA 3' end processing through p21, NPAT and CDK9. Oncogene. 2010;29:2853-63.
- [CrossRef] [PubMed] [Google Scholar]
- Downregulation of the tumor suppressor HSPB7, involved in the p53 pathway, in renal cell carcinoma by hypermethylation. Int J Oncol. 2014;44:1490-8.
- [CrossRef] [PubMed] [Google Scholar]
- Transcriptional factors Eaf1/2 inhibit endoderm and mesoderm formation via suppressing TGF-ß signaling. Biochim Biophys Acta Gene Regul Mech. 2017;1860:1103-16.
- [CrossRef] [PubMed] [Google Scholar]
- Overexpression of CDK2 is a prognostic indicator of oral cancer progression. Jpn J Cancer Res. 2001;92:352-60.
- [CrossRef] [PubMed] [Google Scholar]
- GABPA inhibits invasion/metastasis in papillary thyroid carcinoma by regulating DICER1 expression. Oncogene. 2019;38:965-79.
- [CrossRef] [PubMed] [Google Scholar]
- ENL: Structure, function, and roles in hematopoiesis and acute myeloid leukemia. Cell Mol Life Sci. 2018;75:3931-41.
- [CrossRef] [PubMed] [Google Scholar]
- Expression of KLF9 in pancreatic cancer and its effects on the invasion, migration, apoptosis, cell cycle distribution, and proliferation of pancreatic cancer cell lines. Oncol Rep. 2018;40:3852-60.
- [CrossRef] [PubMed] [Google Scholar]
- How is mRNA expression predictive for protein expression? A correlation study on human circulating monocytes. Acta Biochim Biophys Sin (Shanghai). 2008;40:426-36.
- [CrossRef] [PubMed] [Google Scholar]




