
Translate this page into:
Combination of PARPi and anti-PD-L1 therapies in ovarian cancer

*Corresponding author: Dr. Punit Kaur, Associate Director, Precision Therapeutics Proteogenomics Diagnostics Center, Associate Professor, Department of Medicine, Division of Hematology/ Oncology, Eleanor N. Dana Cancer Center, University of Toledo College of Medicine and Life Sciences, Toledo, Ohio, United States. puneet9@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Morand SM, Ngo NT, Mitchell AC, McHugh MA, Mack ST, Kaur P, et al. Combination of PARPi and anti-PD-L1 therapies in ovarian cancer. Int J Mol Immuno Oncol 2023;8:44-50.
Abstract
Ovarian cancer affects one in 72 female patients in America, and nearly half of the females who carry a BRCA1/2 mutation will be diagnosed in their lifetime. At present, treatment options such as immune checkpoint inhibitors (ICI) and poly (ADP-ribose) polymerase inhibitors (PARPi) are effective against ovarian cancer in a subset of the population. In this article, we review important combination therapeutics that maximize the benefits of these agents in as many patients as possible. PARPi targets deoxyribonucleic acid (DNA) repair mechanisms inside malignant cells, inducing cell death through synthetic lethality. ICIs target immunogenic antigens expressed on the surface of malignant cells so that the immune system can eliminate cancer cells. There is a direct relationship between the degree of DNA damage, also known as the tumor mutational burden and the effectiveness of ICIs. This principle suggests that treatments combining PARPi and ICI may allow DNA damage to accumulate by interrupting repair mechanisms, which may result in newly expressed antigens that could be targeted by the bolstered immune system. In addition, heat-shock proteins (Hsps) are upregulated during cellular stress, such as the stress elicited by the immense metabolic demand of cancer cells. Hsp has potential as prognostic biomarkers, and further, study is required to see how they interact with treatment options. More specifically, both Hsp60 and Hsp10 may represent a prognostic biomarker for ovarian cancer, and further, research into their mechanisms is important. ICI and PARPi combinatorial therapies for ovarian cancer may extend the benefits of each drug to a larger population, and Hsp represents an opportunity for predicting outcomes and tracking responses in cancer patients.
Keywords
Anti-programmed death ligand-1
Combination therapy
Heat-shock proteins
Ovarian cancer
Poly (ADP-ribose) polymerase inhibitors
Tumor mutational burden

INTRODUCTION
In the United States, over 22,000 female patients are diagnosed with ovarian cancer each year, and 1.3% of American females will be diagnosed in their lifetime.[1,2] This number increases in females diagnosed with mutations in the infamous BRCA1/2 genes, and females with BRCA1 or BRCA2 mutations have a 44% and 17% chance, respectively, of developing ovarian cancer[3] [Figure 1]. The prognosis for ovarian cancer is highly dependent on the stage at the time of diagnosis, with overall survival of only 45% after 5 years. This number is much more dismal in patients with stage 4 disease, of whom only 17% can be expected to survive 5 years.[1,2] Per the National Comprehensive Cancer Network, the standard of care is composed of a debulking surgery to remove the tumor, followed by platinum-taxane chemotherapy that hopes to eliminate cancer cells at the tumor margins and early metastases. Two targeted therapies include poly (ADP-ribose) polymerase (PARP) inhibition in women carrying the BRCA1/2 mutation and immune-checkpoint inhibitor (ICI) therapies in high microsatellite instability (MSI-H) tumors. In this chapter, we will explore current data on PARP inhibitors (PARPi) and ICIs as a potential combination therapy for ovarian cancer, and we will consider the role that heat-shock proteins may have in the efficacy of treatment.

- The incidence of ovarian cancer in American women. The incidence of ovarian cancer in American women is 1 in 72 (1.3%). For women with BRCA1 mutations, the incidence rises to 32 in 72 (44%).
BRCA1/2
BRCA1/2 is a tumor suppressor gene encoding the proteins BRCA1 and BRCA2, respectively, which are largely involved in deoxyribonucleic acid (DNA) repair mechanisms. They enter multiple pathways including homologous repair (HR) and non-homologous end-joining (NHEJ), where they interact with repair proteins to ensure stability of the genome. When one or both proteins are defective, patients have a higher likelihood of acquiring DNA damage. This damage leads to a buildup of mutations, which allows cells to transform into cancer.[4] Indeed, patients with BRCA1/2 mutations have a higher incidence of breast and ovarian cancers.[3] The BRCA1/2 mutation is present in an estimated 0.2% of the general population and 5–15% of ovarian cancers are found to have mutations in the BRCA1/2 genes.[5] Although the BRCA1/2 mutation is famously known for breast and ovarian cancers, it can be present in any histological cancer, including fallopian tube cancer,[6-8] peritoneal cancer,[9] prostate cancer,[10] and pancreatic cancer.[4,11]
PARPi
PARP is a class of enzymes that includes PARP1, PARP2, and PARP3. These PARP proteins are involved in DNA repair pathways such as base-excision repair (BER), HR, NHEJ, and alternative non-homologous end-joining (Alt-EJ). PARP enzymes catalyze PARylation, the addition of negatively charged PAR molecules onto glutamate, aspartate, or lysine residues, which impacts the way proteins interact with other proteins and DNA [Figure 2]. This is integral to the formation of DNA repair complexes in all the repair pathways.[12,13] PARP proteins have become a target for cancer therapeutics. Mechanistically, PARPi competes with NAD+ at the PARP catalytic domain, effectively blocking PARylation and the subsequent formation of DNA repair complexes. This is effective against cancer cells with a BRCA1/2 mutation by the mechanism of synthetic lethality. Synthetic lethality posits that impairing the DNA repair system at 2 levels results in rapid cell death due to excessive accumulation of DNA damage, rendering the cell incapable of survival. By contrast, a single impairment in DNA repair, such as a BRCA1/2 mutation, does not necessarily result in cell death because there are alternative pathways (such as BER and Alt-EJ) that the cell can use to repair lethal mutations. Thus, PARPi is indicated in patients with BRCA1/2 mutations by the principle of synthetic lethality.[13] A large and randomized phase III trial comparing Olaparib (a PARPi) monotherapy to single-agent chemotherapy in BRCA-mutated ovarian cancer showed significant clinical improvement in the PARPi arm, with an overall response rate of 72% to PARPi compared to 51% with standard chemotherapy. There is some evidence that PARPi alters the immune landscape of the tumor microenvironment with notable changes including increased programmed death ligand-1 (PD-L1) expression by tumor cells, activation of immune pathways, and increased mutation burden secondary to genomic instability.[14]

- The mechanism of Poly (ADP-ribose) polymerase (PARP) inhibitors. (a) shows the normal mechanism of PARP class proteins. PARP proteins have chains of PAR connected by ribosyl links. With NAD+ as a substrate, PARP can transfer PAR onto glutamine, lysine, or arginine residues on proteins to alter the way they interact with other molecules in the cell. (b) shows PARP inhibitors binding to the substrate site of the PARP protein, which prevents the binding of NAD+ and the subsequent PARylation of cell proteins. (Alsaab et al., 2017).
PD-L1 INHIBITORS
PD-L1 inhibitors are a class of ICIs that enhance innate and adaptive immunity by inhibiting immunosuppressive mechanisms exacted by cancer cells. In healthy tissue, PD-L1 on dendritic or antigen-presenting cells binds to PD receptor-1 (PD-1) on T-cells to inhibit immune activation, cytokine release, and cell elimination [Figure 3]. This protects self-cells against cytotoxic T-lymphocyte-mediated autoimmune reactions. In cancer, tumor cells may present PD-L1 to disable the immune system, control the tumor microenvironment, and prolong tumor survival. ICIs such as anti-PD-L1 have been developed to counter this immune evasive strategy and bolster the immune response to cancer.[15] At present, PD-L1 inhibitors are well studied in melanoma,[16] non-small cell lung cancer,[17] and bladder cancer[18] and they are being studied broadly across cancer histology.[19] In general, ICIs have shown limited response in gynecologic cancers, likely due to a “cold” tumor environment that fails to stimulate the immune system. Some populations of gynecologic cancer patients may receive benefits. In ovarian cancer, ICI therapy such as anti-PD-L1 therapy is indicated in tumors with MSI-H that are refractory to treatment with platinum therapy. Microsatellite instability refers to an impairment in mismatch repair that renders a DNA segment more susceptible to mutation and MSI-H is used as a biomarker predictive of response to ICI therapy.[20]

- The mechanism of programmed death ligand-1 (PD-L1) immune suppression. (a) The purple wedges represent tumor neoantigens, which activate T-cell receptors (purple triangles) on cytotoxic T lymphocytes (CTL) and result in the destruction of the tumor cell by the T-cell. (b) PD-L1 on the cancer cell (green circle) binds to PD-1 on the CTL (green crescent) to evade destruction.
TUMOR MUTATIONAL BURDEN (TMB)
TMB defines the quantity of non-synonymous mutations per megabase of tumor DNA. For each new mutation, the affected protein may or may not expose the altered segment to the cell’s exterior. For example, some mutations may affect an intracellular protein, and some may have no effect on protein structures. In these cases, the exterior of the cancer cell appears immunologically like a non-cancer self-cell. If the mutated protein is exposed, it is considered a neoantigen that may be detected by the immune system. This marks the cell for destruction, given the adequate functioning of the immune system. Recall, however, that the presence of the PD-1/PDL1 interaction may disturb this mechanism. Although not all new mutations yield a neoantigen, the relative quantities of each are related, with higher TMB being associated with more neoantigens.[20] Furthermore, high neoantigen levels and high TMB are both related to better overall survival and enhanced response to immune-checkpoint therapies[21] including PD-L1 inhibitors.[22] Mechanistically, high TMB with multiple neoantigens allow the immune system to “find” cancer cells, and the PD-1/PD-L1 checkpoint inhibition allows the immune system to eliminate them [Figure 4].

- Lymphocytic response to tumor mutational burden (TMB). (a) High-TMB tumors present multiple neoantigens, which can be bound by multiple different populations of circulating T-cells, thereby increasing the likelihood that a T-cell will encounter cancer and mount an immune response. (b) Medium-TMB tumors present less neoantigens. They may still be identified by the immune system, but the likelihood is lower. (c) Low-TMB tumors present almost no neoantigens. This makes it difficult for T-cells to identify the cancer as non-self.
HEAT-SHOCK PROTEINS (Hsps)
To carry out normal functions, human cells depend on the proper transcription, translation, and folding of proteins. Essential to this process are Hsps, a class of highly conserved, ubiquitous, and constitutively expressed chaperone proteins. Although they derive their name from the heat shock/stress process, during which Hsp is highly expressed, Hsp is constitutively present at low levels in all cells. Hsps have an integral role in the folding of a single protein and the assembly of a multiprotein complex[23] [Figure 5]. In this sense, Hsp has a role in a wide breadth of cellular functions, including protein homeostasis, apoptosis, angiogenesis, cell cycle control, and growth factor signaling. Notably, many of these functions are enhanced in cancer and have been identified as cancer hallmarks.[24] Similarly, the rapid growth of cancer cells creates a state of cellular stress, which ushers in the upregulation of Hsp. Therefore, Hsp is an interesting correlate of cancer.
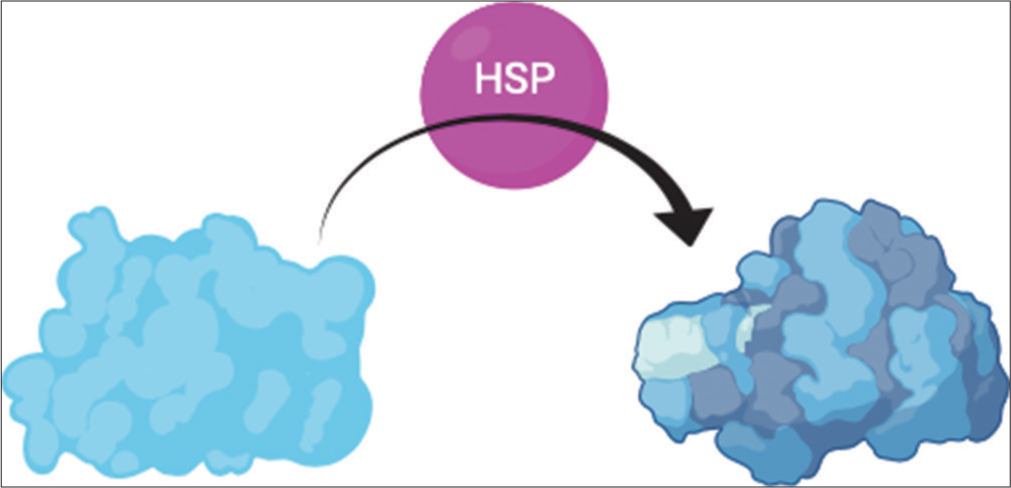
- Heat shock proteins (Hsps) facilitate the folding of proteins into their tertiary structure and the aggregation of multiprotein complexes. Hsps interact with translated proteins to facilitate folding into the correct tertiary structure. The protein to the left represents the initial folding due to thermodynamic factors such as hydrophobicity, and the protein to the right shows the final tertiary structure, which was achieved with the help of Hsp.
It was originally believed that Hsp functions exclusively inside the cells. It has been discovered that several family members of Hsp, including Hsp60 and Hsp72, exist extracellularly in times of cellular stress, such as after necrosis or mild secretion.[24-30] In this setting, Hsp, particularly Hsp72, may be associated with infection or disease, where they can signal that a threat is present, thereby playing a role in immunity.[31] Hsp72 has two characteristics that enable its role in immunity. First, Hsp72 has a peptide-binding functionality that creates an immunological “fingerprint” of the diseased cells. This “fingerprint” can then be recognized by the immune system as a threat. Antigen-presenting cells accept the chaperoned peptides from Hsp to induce the priming of CD8+ T lymphocytes that will target cells expressing the peptide. This elicits antigen-specific immunity that can recognize the disease-causing cell in the future.[32] In addition, Hsp72 can induce non-specific stimulation of pro-inflammatory cytokine and chemokine production.[32-34] In sum, extracellular Hsp72 influences the ability of the innate and adaptive immune systems to detect and fight disease-causing cells. The ability of Hsp to serve as both a chaperone and a cytokine is now referred to as the “chaperokine” activity of Hsp [Figure 6].
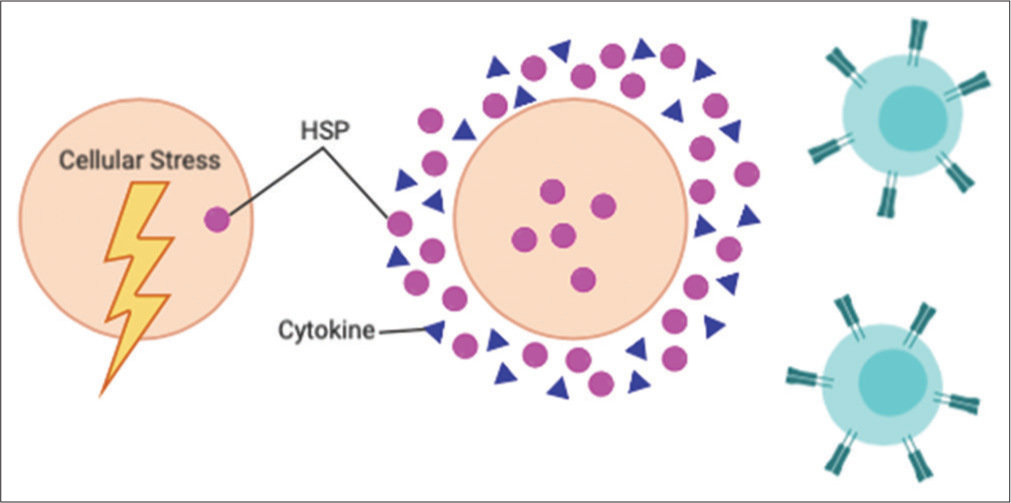
- Heat shock proteins (Hsps) response to cellular stress. In times of cellular stress, Hsps are upregulated within the cell and in the extracellular space. In addition, they aid the innate and adaptive immune system in identifying the source of cellular stress by creating an immunological “fingerprint” of the offending cell. Finally, they encourage the release of proinflammatory cytokines, which augment the immune response.
Although Hsp in ovarian cancer is less well-studied than Hsp in other cancer types, there is emerging evidence that Hsp10 may be a biomarker for ovarian cancer. Hsp10, also known as the “early pregnancy factor,” has several roles in cancer progression. First, it mediates T-cell dysfunction by blocking CD3-zeta, a protein necessary for T-cell activation. Hsp10 was detected in samples from ovarian cancer patient serum, ascites fluid, and tumor cells, but it was undetectable in the control population. Because Hsp10 can be detected in serum samples, it represents a potential biomarker for ovarian cancer that is less expensive than imaging and less invasive than a biopsy. Other Hsp, such as Hsp60, has been identified as prognostic for ovarian cancer. In fact, one study found that Hsp60-high patients with ovarian cancer had a median survival time of 46.8 months compared to a median survival time of just 22.1 months in Hsp60-low patients. These findings are early indications of the utility of Hsp as biomarkers for the diagnosis and prognosis of ovarian cancer.
DISCUSSION
In this chapter, we have discussed the role of PARP inhibition and anti-PD-L1 therapy in ovarian cancer. PARP inhibition promotes DNA damage, thereby promoting the accumulation of new tumor mutations. This drives a higher TMB and larger quantity of neoantigens, which renders a cancer cell more susceptible to detection by the immune system. By bolstering the immune system’s access to cancer cells by blocking PD-1/PD-L1 interactions, ICIs represent a logical addition to PARPi for the treatment of cancer. More studies are required to evaluate the effectiveness of this therapeutic combination. Indeed, studies are currently underway to assess their utility as a combination. Further, combinatory options could include other DNA-damaging agents, such as chemotherapy and radiation, with an immune-bolstering agent like anti-PD-L1. By the above logic, inducing TMB by damaging DNA could enhance immune detection and elimination of cancer cells. In fact, a relationship has been shown between ICIs and various DNA-damaging agents.[35] While promising, this requires further research to define the mechanism and select the optimal combination agents. Due to the poor side effect profiles of chemotherapeutics and radiation therapies, PARPi represents a promising alternative to enhance ICI therapies while minimizing adverse effects. Future work includes verifying this relationship in gynecologic cancers and evaluating the utility of PARPi + ICI therapy in a wider range of cancers.
Finally, HSPs represent an emerging area of interest for the cancer community. HSPs may represent biomarkers of diagnosis and prognosis that can be detected in serum, making them relatively easy to track. In ovarian cancer, Hsp10 has a role in T-cell suppression. In this way, the relationship between Hsp10 and ICI therapies, which enhance T-cell activity, warrants further research. Similarly, understanding the mechanism of why Hsp60 increases survival more than two-fold in Hsp60-high patients compared to Hsp60-low patients will help us design more personalized treatments.
CONCLUSION
This article presents HSP as a potential biomarker for ovarian cancer. Novel single therapeutics warrant the exploration of novel combinatorial therapies as exemplified by PARPi and ICI combination therapy outlined in the article. Matching therapeutics based on mechanisms is a sensible approach in discovering synergism. Since the PARPi binding to PARP at the 5-dRP end, trapping and maintaining the PARP-DNA complex presents a physical obstacle to the replication machinery. It has been reported that in HRR-deficient cancer cells, trapped PARP results in replication fork collapse and finally leads to cell death. Therefore, the pairing of a therapy that negates DNA repair mechanisms (PARPi) with a therapy that targets cells with a high mutational burden (ICI) is a logical combination and indeed suggests a positive therapeutic benefit to this approach. With the rapid development of novel and targeted cancer therapeutics, it is imperative to identify biomarkers to measure the efficacy of novel therapeutics, as well as to distinguish between potential “good responders” and “poor responders” to therapy based on these biomarkers. This distinction spares patients with a low likelihood of response to the time and adversity of inefficacious therapy while ensuring that these therapies get to the patients who will benefit the most. For example, the most common adverse events associated with PARPi treatment in clinical trials include fatigue, anemia, neutropenia, thrombocytopenia, nausea, vomiting, diarrhea, headache, and decreased appetite. Most cancer patients with genetic mutations have been reported for tumor shrinkage with the PARPi rucaparib feeling better as compared with systemic chemotherapy. If a PARPi can be used to lengthen the amount of time that a patient has before starting back on systemic chemotherapy, it might become beneficial. Due to the DNA repair defect, BRCA1/2 deficient tumor cells are more sensitive to PARPi through the mechanism of synthetic lethality. At present, more research is underway to confirm the role of PAPRi targeting PARP in various cancers including ovarian and breast cancer. In conclusion, the discovery of effective biomarkers and the combination of mechanistically synergistic therapeutics present an excellent opportunity for both scientists and oncologists to improve therapeutic options for patients diagnosed with cancer.
Acknowledgments
This work was supported in part by the Medical Summer Research Program (MSRP) (to M.A.M., N.T.N., S.M.M., and S.T.M.), a grant from the Mitchell Family (to A.C.M.) and institutional support from the University of Toledo College of Medicine and Life Sciences (to P.K. and A.A.).
Ethical approval for studies involving humans
This article does not contain any studies with human participants performed by any of the authors.
Ethical approval for studies involving animals
This article does not contain any studies with animals performed by any of the authors.
Declaration of patient consent
Patient’s consent not required as there are no patients in this study.
Conflicts of interest
There are no conflicts of interest.
Financial support and sponsorship
Nil.
References
- Surgical management of mandibular subcondylar fractures under local anesthesia: A proposed protocol. J Oral Maxillofac Surg. 2019;77:1040e1-9.
- [CrossRef] [PubMed] [Google Scholar]
- Effect of topical application of pure honey in chemo-radiation-induced mucositis and its clinical benefits in improving quality of life in patients of oral squamous cell carcinoma. J Maxillofac Oral Surg. 2019;18:73-9.
- [CrossRef] [PubMed] [Google Scholar]
- Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA. 2017;317:2402-16.
- [CrossRef] [PubMed] [Google Scholar]
- BRCA germline mutations in Jewish patients with pancreatic adenocarcinoma. J Clin Oncol. 2009;27:433-8.
- [CrossRef] [PubMed] [Google Scholar]
- The contribution of BRCA1 and BRCA2 to ovarian cancer. Mol Oncol. 2009;3:138-50.
- [CrossRef] [PubMed] [Google Scholar]
- Cancer risk estimates for BRCA1 mutation carriers identified in a risk evaluation program. J Natl Cancer Inst. 2002;94:1365-72.
- [CrossRef] [PubMed] [Google Scholar]
- BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002;62:6997-7000.
- [Google Scholar]
- Salpingo-oophorectomy and the risk of ovarian, fallopian tube, and peritoneal cancers in women with a BRCA1 or BRCA2 mutation. JAMA. 2006;296:185-92.
- [CrossRef] [PubMed] [Google Scholar]
- Fallopian tube and primary peritoneal carcinomas associated with BRCA mutations. J Clin Oncol. 2003;21:4222-7.
- [CrossRef] [PubMed] [Google Scholar]
- Cancer risks among BRCA1 and BRCA2 mutation carriers. Br J Cancer. 2007;96:11-5.
- [CrossRef] [PubMed] [Google Scholar]
- The role of BRCA1 and BRCA2 mutations in prostate, pancreatic and stomach cancers. Hered Cancer Clin Pract. 2015;13:16.
- [CrossRef] [PubMed] [Google Scholar]
- Role of BRCA mutations in cancer treatment with poly(ADP-ribose) polymerase (PARP) inhibitors. Cancers (Basel). 2018;10:487.
- [CrossRef] [PubMed] [Google Scholar]
- Therapeutic applications of PARP inhibitors in ovarian cancer. Biomed Pharmacother. 2020;127:110204.
- [CrossRef] [PubMed] [Google Scholar]
- Combined PARP inhibition and immune checkpoint therapy in solid tumors. Cancers (Basel). 2020;12:1502.
- [CrossRef] [PubMed] [Google Scholar]
- Progress in clinical trials of photodynamic therapy for solid tumors and the role of nanomedicine. Cancers (Basel). 2020;12:2793.
- [CrossRef] [PubMed] [Google Scholar]
- Long-term survival, quality of life, and psychosocial outcomes in advanced melanoma patients treated with immune checkpoint inhibitors. J Oncol. 2019;2019:5269062.
- [CrossRef] [PubMed] [Google Scholar]
- Looking for the best immune-checkpoint inhibitor in pre-treated NSCLC patients: An indirect comparison between nivolumab, pembrolizumab and atezolizumab. Int J Cancer. 2018;142:1277-84.
- [CrossRef] [PubMed] [Google Scholar]
- Immune checkpoint inhibitors for metastatic bladder cancer. Cancer Treat Rev. 2018;64:11-20.
- [CrossRef] [PubMed] [Google Scholar]
- Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int Immunopharmacol. 2018;62:29-39.
- [CrossRef] [PubMed] [Google Scholar]
- The neoepitope landscape of breast cancer: Implications for immunotherapy. BMC Cancer. 2019;19:200.
- [CrossRef] [PubMed] [Google Scholar]
- Genomic correlates of response to immune checkpoint blockade. Nat Med. 2019;25:389-402.
- [CrossRef] [PubMed] [Google Scholar]
- PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4.
- [CrossRef] [Google Scholar]
- HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11:515-28.
- [CrossRef] [PubMed] [Google Scholar]
- Heat shock protein-containing exosomes in midtrimester amniotic fluids. J Reprod Immunol. 2008;79:12-7.
- [CrossRef] [PubMed] [Google Scholar]
- Release of heat shock proteins: Passive vs active release mechanisms In: Asea A, De Maio A, eds. Potent Mediators of Inflammation and Immunity: Heat Shock Proteins. Vol 1. Dordrecht, Netherlands: Springer; 2007. p. :3-20.
- [CrossRef] [Google Scholar]
- Hsp70: A chaperokine In: Henderson B, Pockley AG, eds. The Biology of Extracellular Molecular Chaperones. Vol 291. Chichester, United Kingdom: Novartis Foundation Symposium, Wiley Press; 2007. p. :173-83.
- [CrossRef] [PubMed] [Google Scholar]
- Heat Shock Proteins: Potent Mediators of Inflammation and Immunity. Dordrecht, Netherlands: Springer; 2007. p. :357.
- [CrossRef] [Google Scholar]
- Heat shock proteins in physiology and pathology: The Berlin meeting. Cell Stress Chaperones. 2007;12:205-8.
- [CrossRef] [PubMed] [Google Scholar]
- Regulation of signal transduction by intracellular and extracellular Hsp70 In: Henderson B, Pockley AG, eds. The Extracellular Biology of Molecular Chaperones. Vol 1. London: Cambridge University Press; 2005. p. :133-43.
- [CrossRef] [Google Scholar]
- Novel signal transduction pathway utilized by extracellular HSP70: Role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028-34.
- [CrossRef] [PubMed] [Google Scholar]
- HSP70 peptide-bearing and peptide-negative preparations act as chaperokines. Cell Stress Chaperones. 2000;5:425-31.
- [CrossRef] [PubMed] [Google Scholar]
- HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med. 2000;6:435-42.
- [CrossRef] [PubMed] [Google Scholar]
- Trial watch: The clinical trial landscape for PD1/PDL1 immune checkpoint inhibitors. Nat Rev Drug Discov. 2018;17:854-5.
- [CrossRef] [PubMed] [Google Scholar]
Co-Corresponding Author:

Dr. Alexzander Asea, President and CEO, NampEVA BioTherapeutics LLC, Dover, DE - 19901, United States and Professor of Medicine and Director, Precision Therapeutics Proteogenomics Diagnostics Center, Department of Medicine, Division of Hematology/Oncology, Eleanor N. Dana Cancer Center, University of Toledo College of Medicine and Life Sciences, 3000 Arlington Avenue, Toledo, OH - 43614, United States. president@nampeva.com




